Mutacja, która wystąpiła u danej osoby, powodując chorobę mitochondrialną, może być mutacją de novo. Jednak w większości przypadków jest ona odziedziczona od rodziców, którzy są nosicielami wadliwego genu. Jest to ważna informacja w przypadku planowania potomstwa i ryzyka wystąpienia choroby przez przyszłe dzieci, oraz możliwości przekazania mutacji w obrębie dalszej rodziny – wujków, ciotek, kuzynostwa – którzy również mogą okazać się nosicielami konkretnej mutacji.
Należy pamiętać, że choroby mitochondrialne, mogą być spowodowane zarówno przez DNA jądrowe (nDNA) jak i DNA mitochondrialne (mtDNA). W przypadku mtDNA zachodzi inny sposób dziedziczenia niż w przypadku nDNA. Różne są również sposoby i możliwości diagnostyki prenatalnej w przypadku obu genomów.
Typy dziedziczenia chorób mitochondrialnych obejmują:
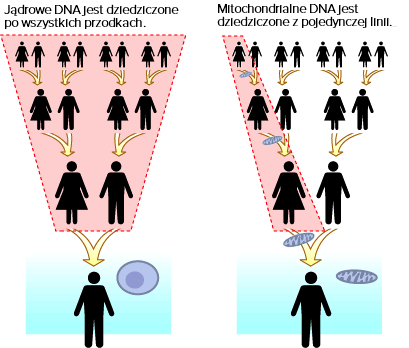
DZIEDZICZENIE DNA JĄDROWEGO (nDNA) – DZIEDZICZENIE AUTOSOMALNE
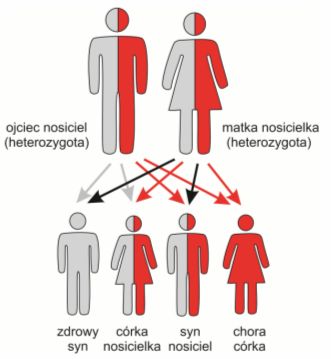
Dziedziczenie autosomalne recesywne (każdy z rodziców jest nosicielem zmutowanego genu)
Choroba dziedziczona autosomalnie recesywnie jest efektem uszkodzenia obu kopii (alleli) genu. Oznacza to, że obydwoje rodzice chorego dziecka muszą być bezobjawowymi nosicielami uszkodzonej kopii, co więcej muszą mu tę kopie przekazać. Ryzyko ujawnienia się choroby u dziecka wynosi wówczas 25%, niezależnie, czy dziecko jest chłopcem czy dziewczynką. Nie ma też znaczenia liczba posiadanych już dzieci, ryzyko to wciąż jest takie samo.
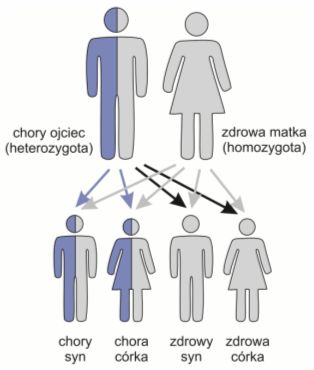
Dziedziczenie autosomalne dominujące (jeden z rodziców jest nosicielem zmutowanego genu)
Choroba dziedziczona autosomalnie dominująco ujawnia się, gdy uszkodzona jest tylko jedna kopia danego genu – tylko jeden z rodziców jest nosicielem. Rodzic będący nosicielem mutacji genetycznej dziedziczonej w ten sposób ponosi 50% ryzyko, że jego dziecko będzie chore.
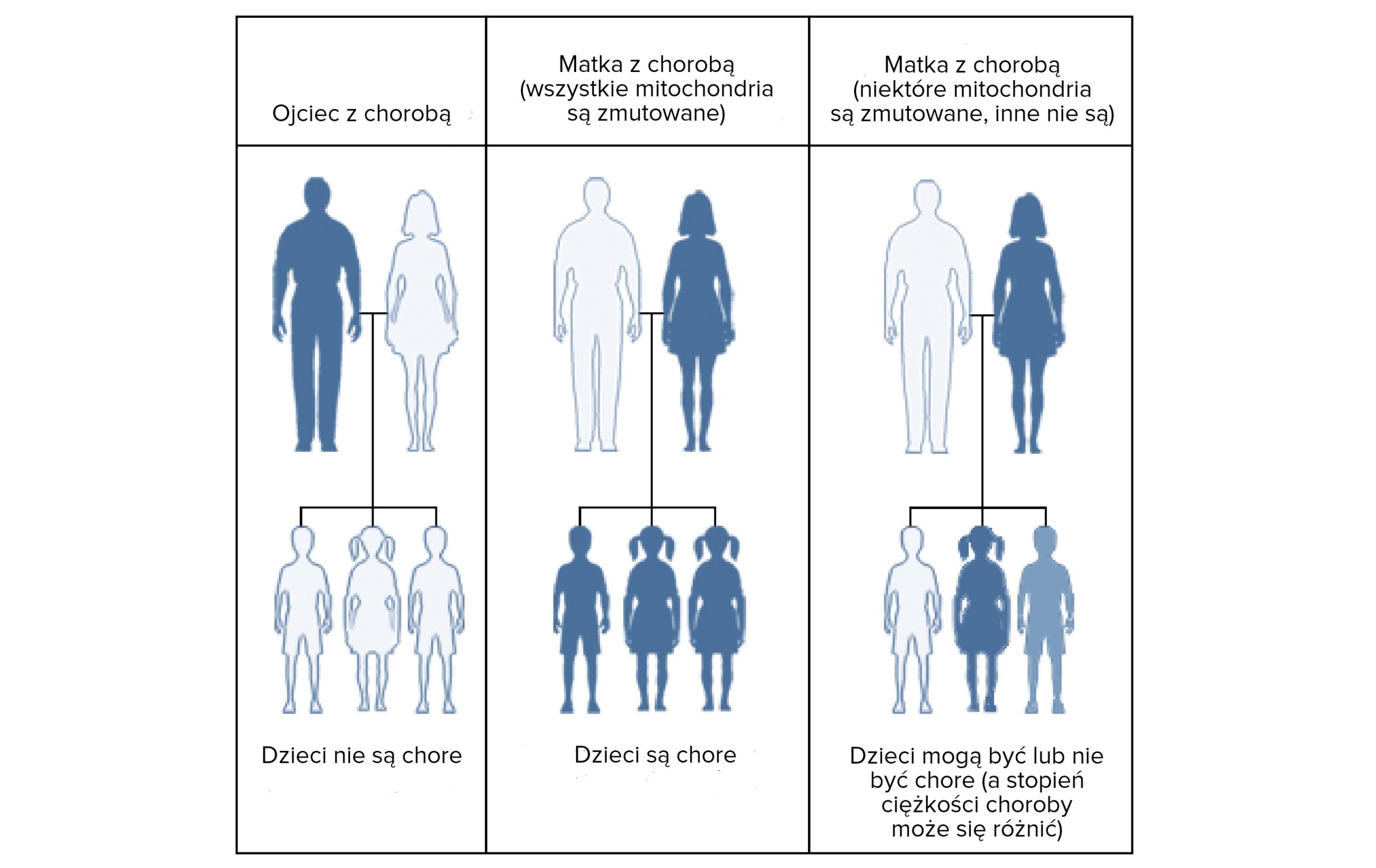
Dziedziczenie DNA mitochondrialnego (mtDNA) – Dziedziczenie mitochondrialne
Dziedziczenie mitochondrialne to sposób dziedziczenia, w którym choroba przekazywana jest wyłącznie przez matkę. Kobieta ze zdiagnozowaną chorobą mitochondrialną przekazuje uszkodzony gen wszystkim swoim dzieciom bez względu na płeć. Dzieci chorego mężczyzny są z kolei zdrowe.
Ryzyko powtórnego wystąpienia choroby i diagnostyka prenatalna
Diagnostyka prenatalna
Wiedząc, że w rodzinie wystąpiła wcześniej mutacja genetyczna, prowadząca do choroby mitochondrialnej, kobieta w czasie ciąży może zostać skierowana na szczegółową diagnostykę prenatalną. Jest ona zwykle wykonywana przy użyciu jednej z dwóch technik: biopsji kosmówki (CVS) lub amniopunkcji i polega na pobraniu komórek lub płynu owodniowego podczas ciąży w celu zbadania znanej mutacji mitochondrialnej. Wynik takiego badania jest analizowany przez genetyka.
Biopsja kosmówki (CVS) jest zwykle wykonywane około 11-12 tygodnia ciąży i polega na pobraniu małej ilości materiału do badania z rozwijającego się łożyska. Kosmówka to najbardziej zewnętrzna błona płodowa, której kosmki są częścią łożyska. Pobranie próbki tych kosmków umożliwia uzyskanie materiału genetycznego płodu w celu sprawdzenia, czy są one nosicielami mutacji mitochondrialnej.
Amniopunkcja jest zwykle wykonywana około 15-18 tygodnia ciąży i polega na pobraniu próbki płynu owodniowego w celu sprawdzenia, czy komórki przenoszą mutację mitochondrialną.
W przypadku mutacji jądrowego DNA zarówno CVS, jak i amniopunkcja mogą bardzo dokładnie określić, czy dziecko jest nosicielem wadliwego genu, a zatem mogą dokładnie określić ryzyko odziedziczenia przez dziecko choroby mitochondrialnej.
W przypadku mutacji mitochondrialnego DNA jest to nieco bardziej skomplikowane. ponieważ ciężkość choroby zależy od liczby wadliwych mitochondriów lub stopnia heteroplazmii Stopień heteroplazmii dla mutacji mtDNA może być różny w różnych organach. W diagnostyce prenatalnej, polegajacej na analizie materiału genetycznego zawartego w komórkach kosmówki, pojawia się problem czy stopień heteroplazmii mierzony w kosmkach kosmówki jest również reprezentatywny dla całego płodu. Dodatkowo czynnikiem utrudniającym ocenę wyniku tego badania i rokowania co do dalszego rozwoju dziecka jest różna tolerancja poszczególnych osób na noszoną mutacją oraz jej stopień (heteroplazmię), i poziom przy którym stopień mutacji powoduje problematyczne objawy. Dwie osoby z tą samą mutacją mitochondrialną i 70% hetroplazmii mogą mię zupełnie rózną skale i obszar symptomów. Jedna z nich może mieć poważne problemy ze stronu ośrodkowego układu nerwowego czy np. poważne problemy kardiologiczne, podczas gdy druga będzie się ” jedynie” szybciej męczyć. W takich przypadkach z pewnością ważne jest, aby zrozumieć wyniki CVS w świetle wpływu na innych osoby w rodzinie oraz poziomu heteroplazmii, w jakim zostały one dotknięte.
Genetyczna diagnostyka przedimplantacyjna (PGD)
Innym sposobem, na stwierdzenie, czy potomstwo będzie dziedziczyło mutacji mitochondrialną, jest diagnostyka przedimplatacyjna (PGD). Jest ona powiązana z technologią in vitro. Badania preimplantacyjne służą diagnostyce w kierunku występowania wad genetycznych oraz mutacji genów w zarodkach, które powstały w wyniku pozaustrojowego zapłodnienia in vitro, zanim dokona się ich transferu do organizmu przyszłej matki.
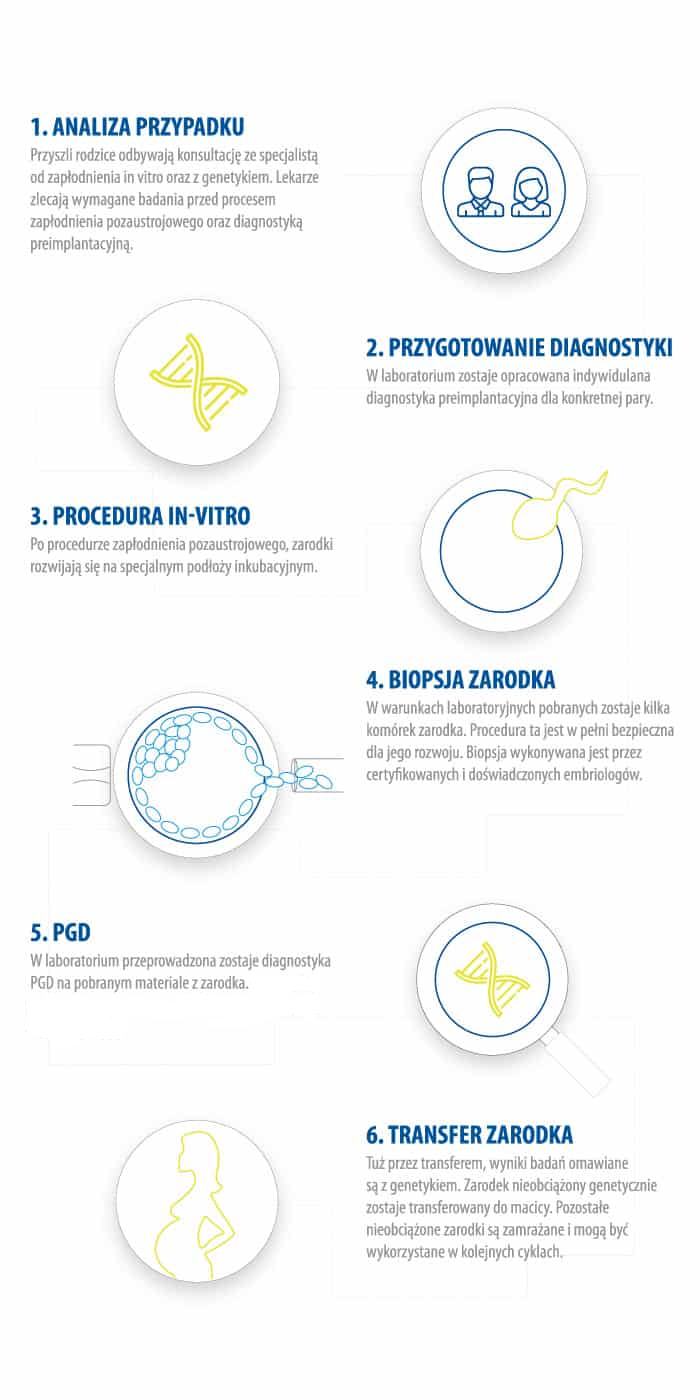
W przypadku mutacji jądrowego DNA, PGD może bardzo dokładnie określić, czy zarodek jest nosicielem wadliwego genu, a więc tylko nienaruszone zarodki zostaną z powrotem wszczepione matce, aby kontynuować ciążę.
Ponownie w przypadku mutacji mitochondrialnego DNA jest to nieco bardziej skomplikowane, ponieważ ciężkość choroby zależy od liczby wadliwych mitochondriów lub stopnia heteroplazmii. Obecność patogennej mutacji mtDNA nie jest jednak równoznaczna z wystąpieniem choroby. Objawy kliniczne wystąpią, jeżeli stopień heteroplazmii przekroczy próg krytyczny, który jest różny dla różnych tkanek – tym niższy, im wyższe zapotrzebowanie energetyczne komórek i tkanek. Zjawisko to nosi nazwę efektu progowego i jest znamienne dla patologii mitochondrialne. W tym samym zespole chorobowym uwarunkowanym mutacją mtDNA stopień heteroplazmii może być inny u poszczególnych chorych, nawet w tej samej rodzinie, i różnić się między tkankami. Co za tym idzie ocena zarodka i rokowanie dalszego prawdłowego rozwoju dziecka jest znacznie utrudnione.