Diagnoza
Choroby mitochondrialne dotyczą tkanek o wysokim zapotrzebowaniu energetycznym i najczęściej objawiają się zajęciem ośrodkowego układu nerwowego (OUN), mięśni szkieletowych, mięśnia sercowego, narządów dokrewnych, nerek, szpiku itd. Za podejrzeniem takiej choroby może przemawiać jednoczesne zajęcie wielu narządów i układów. Zwykle zajęte są OUN i mięśnie, zwłaszcza proksymalne, dlatego choroby te niekiedy określa się jako encefalomiopatie mitochondrialne.
Do najważniejszych objawów potwierdzających Zespół Leigha należą:
- W większości wczesne występowanie objawów w okresie noworodkowym i wczesnodziecięcym( najczęściej pomiędzy 3 miesiącem a 2 rokiem życia). Znane są także przypadki pojawienia się pierwszych symptomów u dzieci przedszkolnych, wczesnoszkolnych, nastolatków a nawet dorosłych, przy wcześniejszym zupełnie prawidłowym rozwoju psychofizycznym.
- Neurodegeneracja powiązana najczęściej z postępującym regresem psychomotorycznym, częstymi dekompresjami organizmu towarzyszącymi najczęściej infekcjom
- Obserwowanie w rezonansie magnetycznym symetrycznych obustronnych zmian w obszarze zwojów jąder podstawy oraz/lub pnia mózgu
- Zauważalne zaburzenia w gospodarce energetycznej organizmu, męczliwość nieproporcjonalna do wysiłku
- Identyfikacja mutacji genetycznej związanej z charakterystycznym genem dla tej choroby
Pomimo coraz większej wiedzy na temat ludzkiego DNA i coraz lepszej dostępności do badań , zdarza się że pomimo charakterystycznych objawów ustalenie mutacji genetycznej jest bardzo trudne lub czasem niemożliwe. Wówczas możliwe jest jeszcze potwierdzenie diagnozy poprzez biopsje mięśnia. Można spotkać się też z określeniami takimi jak: Leigh-Like lub Leigh Spektrum
Zespół Leigha diagnozuje się w stosunku 1:40 000 urodzonych dzieci, bez przewagi płci, a więc zarówno u chłopców jak i dziewczynek. Zespół Leigha rozwija się już w czasie ciąży wraz z rozwojem DNA u zarodka.
Przebieg choroby
Przebieg kliniczny chorób mitochondrialnych z reguły ma charakter postępujący. W różnych okresach choroby mogą pojawiać się objawy z narządów wcześniej nie zmienionych chorobowo lub zaostrzać się objawy już występujące.
Choroby wynikające z deficytów łańcucha oddechowego przebiegają z okresami pogorszeń podczas infekcji, silnych stresów emocjonalnych, zabiegów operacyjnych, a następnie okresami względnej poprawy. W okresach dobrostanu wartości biochemiczne (kwas mlekowy, pirogronianowy, amoniak) mogą być prawidłowe. Po okresach dekompensacji możliwy jest powrót do stabilnej sytuacji, jednak niemożliwy jest powrót do tego samego stanu, który prezentowany był przed dekompensacją.
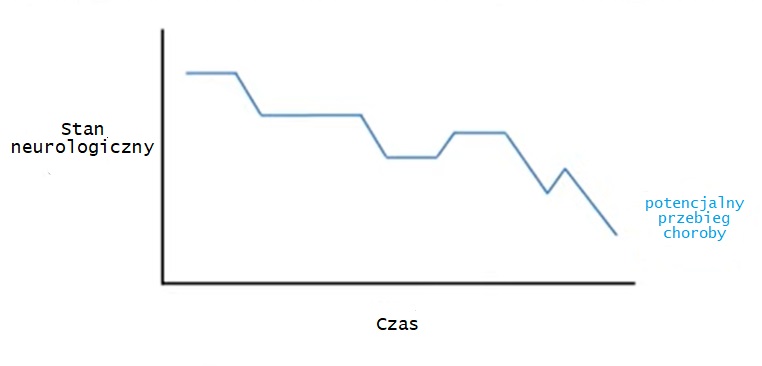
W przypadku układu nerwowego obserwuje się różne postępujące zespoły neurologiczne, w tym: udary w młodym wieku, oftalmoplegię zewnętrzną z ptozą, izolowaną głuchotę sensoryczną, neuropatię obwodową, padaczkę, otępienie lub regres rozwoju. Występują też migrenowe bóle głowy.
Zmiany dotyczące innych narządów obejmują kardiomiopatię, tubulopatię proksymalną z aminoacidurią czy endokrynopatie (cukrzyca, niedoczynność przytarczyc i/lub tarczycy, niedobór hormonu wzrostu. Pojawiającym się zaburzeniom funkcji poznawczych towarzyszą niekiedy różne zaburzenia psychiczne, zazwyczaj: depresja, zaburzenia rzekomonerwicowe, dysforia, znacznie rzadziej epizody psychotyczne urojeniowo-omamowe i schizofrenopodobne. Dzieci prezentują często zaburzenia lękowe.
Szczegółowe symptomy, które mogą się pojawić ze strony poszczególnych układów, omówione zostały w zakładce OBJAWY.