Mutacje DNA a choroby mitochondrialne
Każda komórka w ciele ma jądro w którym zawarta jest informacja genetyczna -DNA, ale oprócz tego w każdej komórce obok jądra komórkowego znajdują się mitochondria. Jest ich około 10-20 w zależności od typu komórek. Mitochondria to niesamowite organelle, która jeszcze 1,5 bilion lat temu były bakteriami, a pozostałością po tym jest posiadanie przez każde mitochondrium własnej informacji genetycznej nazywanej mtDNA. W związku z tym, aby komórki, tkanki, organy mogły prawidłowo funkcjonować muszą zsynchronizować się dwa sprawne genomy: DNA zawarte w mitochondriach (który dziedziczony jest tylko po matce) oraz DNA jądrowe (dziedziczone od obojga rodziców).
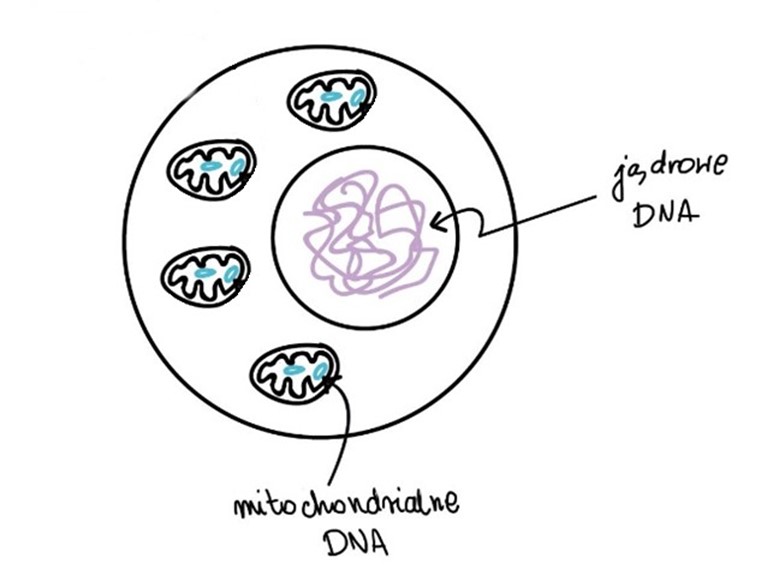
mt-DNA = DNA mitochondrialne n-DNA = DNA jądrowe
Mitochondrii w komórce jest od kilku do kilkunastu, a każde z nich ma swoje odrębne mtDNA. Może się więc zdążyć, że w jednej komórce część mitochondrii ma zmutowane DNA, a część prawidłowe. Czasami ilość uszkodzonych mitochondriów jest na tyle mała, że pozostałe są w stanie poradzić sobie w sprawnym funkcjonowaniu komórki. Jednak zwykle kiedy ilość mitochondriów posiadających zmutowane mtDNA przewyższa ilość nieuszkodzonych mitochondriów, komórka nie jest w stanie efektywnie pracować. Można wówczas zaobserwować poważne konsekwencje m.in. neurologiczne, kardiologiczne. Dlatego w przypadku chorób mitochondrialnych można spotykać się z pojęciami heteroplazmii (tylko pewien procent mitochondrii w komórce ma uszkodzone mtDNA np.60%) lub homoplazmii (wszystkie mitochondria w komórce mają uszkodzone mtDNA – 100%).
Choroby mitochondrialne, które będą prowadziły do zaburzeń funkcjonowania mitochondriów wiążą się z mutacją genetyczna w obrębie albo DNA mitochondrialnego, albo jądrowego. U przeważającej liczby osób chorujących na choroby mitochondrialne stwierdza się mutację w obrębie DNA jądrowego. Jedynie u ok.20% chorych stwierdza się mutacje w mtDNA. Pozostałe przypadki, z mutacją w DNA jądrowym, powiązane są z genami recesywnymi występującymi u obojga rodziców.

Upośledzony proces oddychania komórkowego w chorobach mitochondrialnych
Mitochondria nazywane są często „elektrowniami komórkowymi” ponieważ pełnią w organizmie bardzo ważną rolę w procesie zapewnienia organizmowi potrzebnej mu do fukcjonowania energii. W tym celu produkowana jest wysokoenetgetyczna cząsteczka ATP (adezynotrifosforan).
Zamiana spożywanego jedzenia w energie, odbywa się na poziomie każdej pojedynczej komórki w procesie nazywanym oddychaniem komórkowym. Składa się on z czterech etapów, z których oprócz pierwszego trzy pozostałe mają miejsce we wnętrzu mitochondrium. Szczegółowy przebieg tego mechanizmu można zobaczyć w poniższym nagraniu:
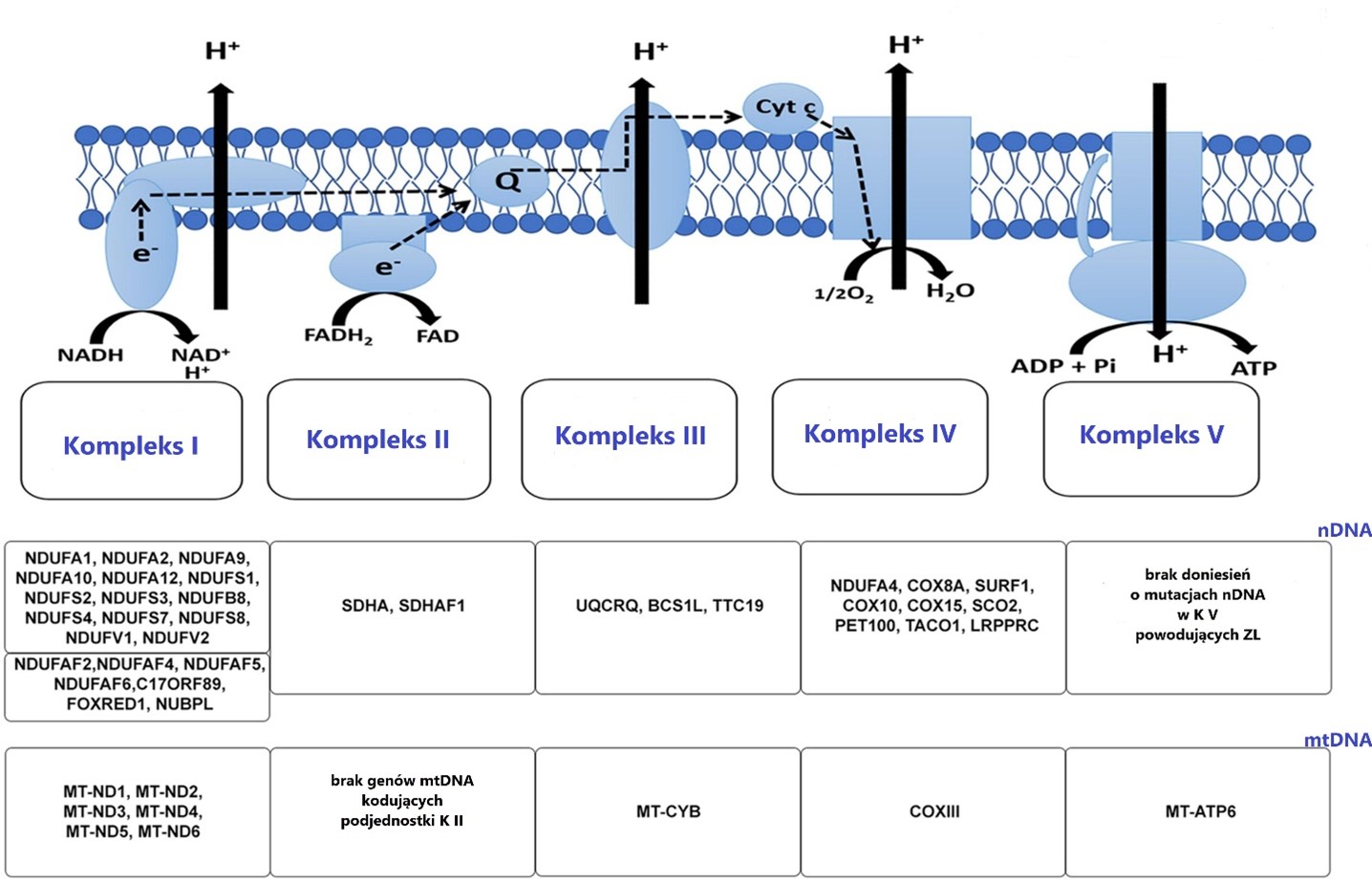
W kontekście chorób mitochondrialnych, szczególnie ważny jest ostatni proces oddychania komórkowego: fosforylacja oksydacyjna/ łańcuch elektronowy. To właśnie na tym etapie produkowane jest najwięcej cząsteczek energetycznych ATP. W tym celu szereg procesów chemicznych powinien przejść przez płynnie przez pięć kompleksów łańcucha oddechowego. Mutacje zarówno w DNA mitochondrialnym, jak i jądrowym utrudniają, a często uniemożliwiają efektywne przejście cząsteczek przez kolejne etapy łańcucha. Na podstawie badań zauważono które mutacje wpływają na podjednostki poszczególnych kompleksów. Z tego względu można się często spotkać z powiązaniem konkretnej mutacji z kompleksem oddechowym, którego dysfunkcje powoduje.
Poniżej najczęściej występujące mutacje powodujące Zespół Leigha ze wskazaniem na łańcuch oddechowy, którego dysfunkcje powodują.